
Terapia de reemplazo enzimático por 15 años en enfermedad de Pompe: reporte de caso
Barra lateral del artículo
Contenido principal del artículo
Resumen
Antecedentes: La enfermedad de Pompe o enfermedad de almacenamiento de glucógeno tipo 2 (OMIM #232300), es un trastorno metabólico hereditario causado por la mutación del gen GAA ubicado en el cromosoma 17q25.3, que afecta la producción de α-glucosidasa ácida, lo que conduce a la acumulación de glucógeno en los lisosomas, especialmente en el músculo estriado y cardiaco, desencadenando múltiples signos y síntomas de presentación variable.
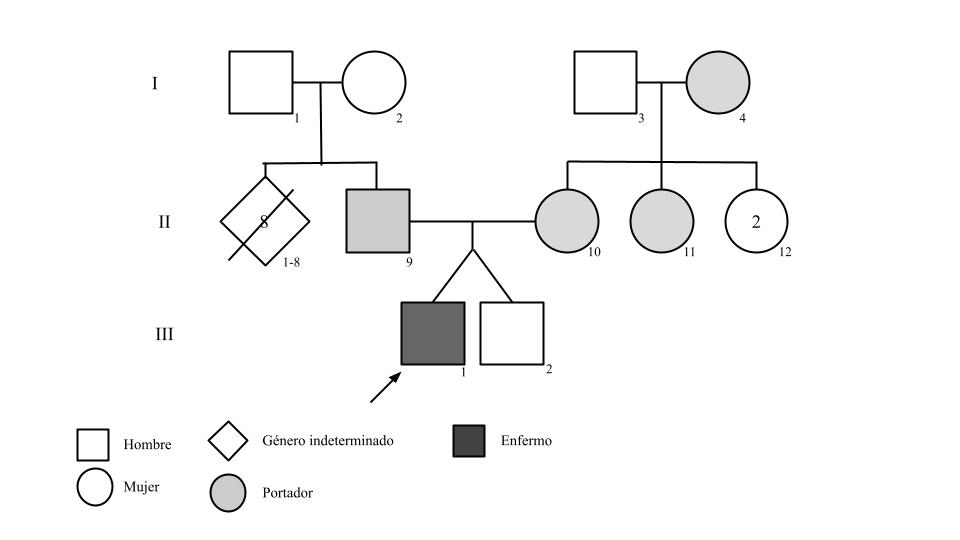
Resumen del caso: El presente caso es de un paciente de 22 años con antecedentes de parto pretérmino y bajo peso, antecedentes familiares de mutación del gen GAA en sus padres portadores y hermano mellizo no portador de esta. Desde los 18 meses de edad, el paciente experimentó dificultad para alimentarse, debilidad en los músculos faciales, cianosis intermitente y síntomas respiratorios progresivos. Adicionalmente, deposiciones líquidas diarias y anomalías hepáticas. Después de una serie de eventos adversos, se confirmó el diagnóstico de glucogenosis y se inició tratamiento con terapia de reemplazo enzimático, lo que resultó en mejoría de los síntomas. Actualmente, se mantiene estable con leve compromiso muscular y respiratorio.
Conclusiones: Esta condición tiene baja prevalencia y su presentación y gravedad varían. Para su diagnóstico se requiere sospecha clínica y pruebas de diagnóstico como análisis enzimático y secuenciación génica. El manejo integral involucra varios especialistas y la terapia de reemplazo enzimático es el tratamiento principal, aunque la terapia génica muestra avances prometedores.
Descargas
Detalles del artículo

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-SinDerivadas 4.0.
Licencia Creative Commons
Atribución-NoComercial-CompartirIgual 4.0 Internacional (CC BY-NC-SA 4.0)
Usted es libre de:
Compartir - copiar y redistribuir el material en cualquier medio o formato.
Adaptar - remezclar, transformar y construir a partir del material
La licencia no puede revocar estas libertades en tanto se sigan los términos de la licencia.
- Atribución — Usted debe dar crédito de manera adecuada, brindar un enlace a la licencia, e indicar si se han efectuado cambios. Puede hacerlo en cualquier forma razonable, pero no de forma tal que sugiera que usted o su uso tienen el apoyo del licenciante.
- NoComercial — Usted no puede hacer uso del material con propósitos comerciales.
- CompartirIgual— Si remezcla, transforma o crea a partir del material, debe distribuir su contribución bajo la misma licencia del original.
- No hay restricciones adicionales — No puede aplicar términos legales ni medidas tecnológicas que restrinjan legalmente a otras a hacer cualquier uso permitido por la licencia.




Referencias
- Sidorina A, Catesini G, Levi Mortera S, Marzano V, Putignani L, Boenzi S, et al. Combined proteomic and lipidomic studies in Pompe disease allow a better disease mechanism understanding. J Inherit Metab Dis. 2021 May;44(3):705-717. https://www.doi.org/10.1002/jimd.12344.
- omim.org. GLUCOSIDASE, ALPHA, ACID; GAA (*606800). https://www.omim.org/entry/606800. (Fecha de consulta: 17 de Abril, 2023)
- Marques JS. The Clinical Management of Pompe Disease: A Pediatric Perspective. Children. 2022 Sep 16;9(9):1404. https://www.doi.org/10.3390/children9091404
- Fatehi F, Ashrafi MR, Babaee M, Ansari B, Beiraghi Toosi M, Boostani R, et al. Recommendations for infantile-onset and late-onset Pompe disease: An Iranian consensus. Frontiers in Neurology. 2021;12. https://doi.org/10.3389/fneur.2021.739931
- Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child. 2020 Oct 2;24(2):3–8 https://doi.org/10.34763/jmotherandchild.20202402si.2001.000002
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Vol. 24, Current Treatment Options in Neurology. Springer; 2022. p. 573–88 .https://doi.org/10.1007/s11940-022-00736-1
- Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Vol. 15, Neurotherapeutics. Springer New York LLC; 2018. p. 928–42. https://doi.org/10.1007/s13311-018-0655-y.
- Taverna S, Cammarata G, Colomba P, Sciarrino S, Zizzo C, Francofonte D, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020 Aug 3;12(15):15856-15874. https://doi.org/10.18632/aging.103794
- Marisa Wexler MS. Pompe disease [Internet]. Pompe Disease News. 2022 [citado el 22 de abril de 2023]. Disponible en: https://pompediseasenews.com/what-is-pompe-disease/
- Unnisa Z, Yoon JK, Schindler JW, Mason C, van Til NP. Gene therapy developments for Pompe disease. Biomedicines [Internet]. 2022;10(2):302. Disponible en: http://dx.doi.org/10.3390/biomedicines10020302
- Sarah B, Giovanna B, Emanuela K, Nadi N, Josè V, Alberto P. Clinical efficacy of the enzyme replacement therapy in patients with late-onset Pompe disease: a systematic review and a meta-analysis. J Neurol [Internet]. 2022 [citado el 22 de abril de 2023];269(2):733–41. Disponible en: http://dx.doi.org/10.1007/s00415-021-10526-5
- Lecis M, Rossi K, Guerzoni ME, Mariotti I, Iughetti L. Enzyme replacement therapy (ERT) on heart function changes the outcome in patients with infantile-onset Pompe disease: A familial history. Case Rep Pediatr [Internet]. 2023 [citado el 23 de abril de 2023];2023:8470341. Disponible en: https://www.hindawi.com/journals/cripe/2023/8470341/
- Kohler L, Puertollano R, Raben N. Pompe disease: From basic science to therapy. Neurotherapeutics [Internet]. 2018 [citado el 23 de abril de 2023];15(4):928–42. Disponible en: https://pubmed.ncbi.nlm.nih.gov/30117059/
- Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child [Internet]. 2020;24(2):3–8. Disponible en: http://dx.doi.org/10.34763/jmotherandchild.20202402si.2001.000002
- Lecis M, Rossi K, Guerzoni ME, Mariotti I, Iughetti L. Enzyme replacement therapy (ERT) on heart function changes the outcome in patients with infantile-onset Pompe disease: A familial history. Case Rep Pediatr [Internet]. 2023 [citado el 23 de abril de 2023];2023:8470341. Disponible en: https://www.hindawi.com/journals/cripe/2023/8470341/